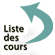6
-
Les pneumopathies interstitielles diffuses de cause inconnue
6
.
1
-
Les granulomatoses
6
.
1
.
1
-
La sarcoïdose
Voir Item 124.
6
.
1
.
2
-
L'histiocytose langerhancienne pulmonaire (anciennement histiocytose X)
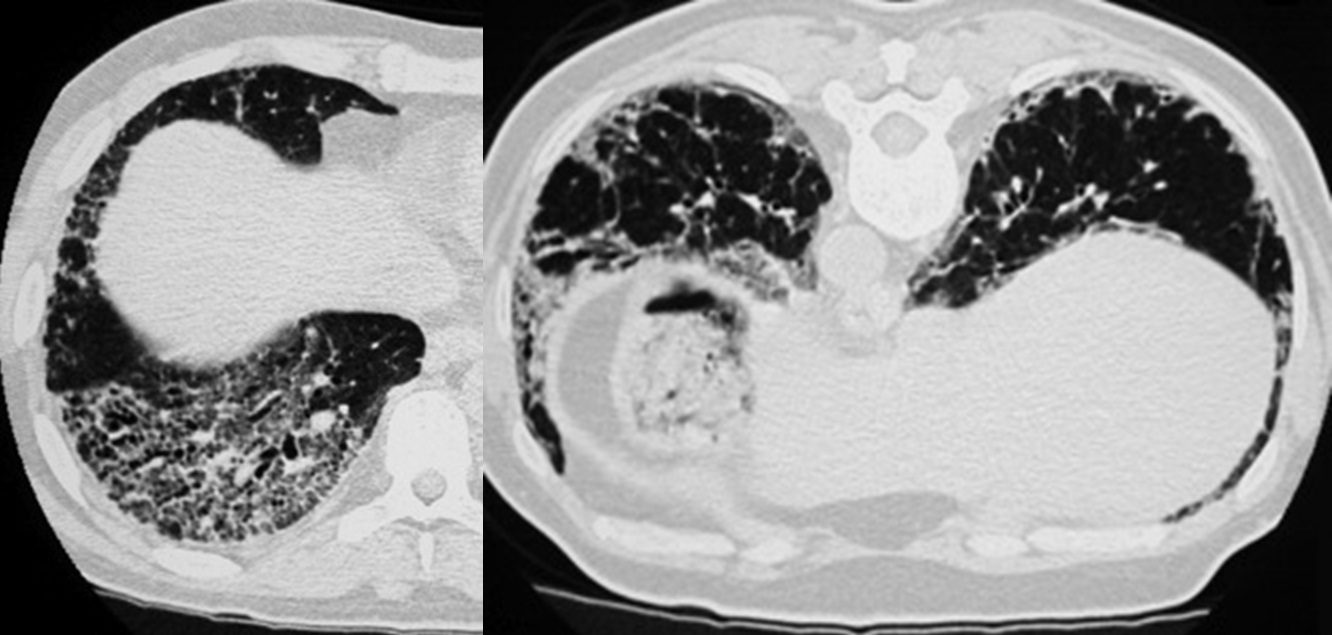
C'est une affection relativement rare, touchant de manière préférentielle l'homme jeune, grand fumeur. Elle se traduit par une toux sèche, une dyspnée à l'effort, parfois des signes généraux, des manifestations systémiques (diabète insipide, granulome éosinophile osseux, atteinte cutanée). Elle peut se compliquer de pneumothorax dans 10 à 20 % des cas. L'aspect tomodensitométrique est très évocateur avec la présence de nodules mal limités, parfois cavitaires et des kystes (à un stade évolué). Les anomalies respectent en général les bases pulmonaires. L'exploration fonctionnelle respiratoire montre un trouble ventilatoire obstructif, une chute du coefficient de transfert du monoxyde de carbone et une désaturation d'exercice. Il existe une alvéolite macrophagique et de nombreuses cellules de Langerhans en immunohistochimie (cellules CD1a positives). La biopsie pulmonaire n'est pas toujours utile tant l'aspect tomodensitométrique est évocateur. L'évolution se fait sur un mode chronique pouvant évoluer dans 10 à 20 % des cas vers une insuffisance respiratoire et une transplantation pulmonaire a parfois été réalisée dans ce contexte.
6
.
2
-
Les pneumopathies interstitielles diffuses idiopathiques
Elles ont longtemps été rassemblées sous le terme de fibrose interstitielle diffuse. On sait aujourd'hui qu'elles regroupent différentes entités, associées à des aspect radiologiques, tomodensitométriques, histologiques différents, avec également des pronostics très variables. L'analyse histologique, nécessite un prélèvement pulmonaire biopsique large pour une analyse assez extensive de l'architecture pulmonaire et de ses éventuels remaniements. Dans ce contexte, les biopsies pulmonaires transbronchiques ne doivent pas être réalisées car elles ne sont pas informatives.
La fibrose pulmonaire idiopathique ou pneumopathie interstitielle commune ou UIP (Usual Interstitial Pneumonia) C'est la plus fréquente des pneumopathies interstitielles diffuses idiopathiques (60 % des cas). Elle débute en général vers 50-70 ans avec une dyspnée progressivement croissante, une toux sèche. L'examen clinique retrouve des râles crépitants bilatéraux prédominants aux bases, avec un hippocratisme digital dans 50 % des cas. À un stade avancé de la maladie, s'y associe des signes d'insuffisance ventriculaire droite. L'exploration fonctionnelle respiratoire objective un trouble ventilatoire restrictif avec altération de la diffusion alvéolocapillaire, et une hypoxémie d'exercice puis de repos. Le lavage bronchopulmonaire objective une hypercellularité avec une augmentation des neutrophiles et parfois des éosinophiles. Sur la radiographie pulmonaire il existe des images réticulaires diffuses prédominantes dans les bases. On note une rétraction pulmonaire. La tomodensitométrie thoracique objective des opacités toujours réticulaires prédominantes aux bases, un aspect parfois pseudo-kystique sous-pleural en rayons de miel, des bronchectasies de traction, des opacités en verre dépoli.
Le diagnostic, nécessite une biopsie pulmonaire vidéo-chirurgicale, qui montre un aspect anatomopathologique très caractéristique.
L'évolution est lentement progressive vers l'insuffisance respiratoire chronique. Le traitement, n'est pas très bien codifié. Des corticoïdes, des immuno-suppresseurs (Cyclophosphamide®, Azatioprine®) ont été utilisés largement mais sont en général peu efficace. L'efficacité de l'INTERFERON-g est à l'étude. La médiane de survie est de 3 ans, et la survie à 10 ans est de l'ordre de 10 %.
La pneumopathie interstitielle non spécifique (PINS) ou NSIP, avec ou sans fibrose associée. Elle débute chez des gens un peu plus jeunes (45-50 ans). Les signes généraux sont habituels. Il existe des crépitants bilatéraux dans les champs pulmonaires. L'hippocratisme digital est plus rare. Il existe parfois une connectivite associée (syndrome de Gougerot-Sjögren ou myopathie idiopathique inflammatoire). Les manifestations pulmonaires peuvent alors précéder la symptomatologie systémique de la connectivite. La TDM montre un aspect en verre dépoli et des opacités alvéolaires non systématisées à prédominance péri-broncho-vasculaire. Le LBA montre une hyper cellularité mixte avec habituellement une majoration de la lymphocytose. Une biopsie pulmonaire chirurgicale est nécessaire au diagnostic. La corticosensibilité est de règle mais une évolution vers l'insuffisance respiratoire chronique est décrite surtout dans les formes avec un pattern fibrotique associé. La survie à 10 ans est de l'ordre de 60 à 70 %.
La pneumopathie organisée ou bronchiolite oblitérante avec pneumopathie organisée (BOOP) peut s'observer au cours de nombreuses circonstances (bactériennes, virales, médicamenteuses, connectivites, radiothérapie, ou idiopathiques). L'installation est le plus souvent subaiguë, avec altération de l'état général, toux sèche, dyspnée d'effort. Il existe des opacités alvéolaires migratrices, corticosensibles ou parfois de véritables pneumopathies interstitielles diffuses. Le diagnostic peut être fait en biopsie transbronchique mais nécessite souvent une biopsie pulmonaire vidéo-assistée de plus grande taille. Cette pneumopathie organisée est associée à une forte corticosensibilité avec une régression en quelques semaines des symptômes. Les rechutes sont très fréquemment décrites et la corticothérapie doit souvent être prolongée.
La pneumopathie interstitielle desquamative ou pneumopathie alvéolaire à macrophages ainsi que la bronchiolite respiratoire avec pneumopathie interstitielle sont des formes rares de pneumopathies interstitielles diffuses idiopathiques touchant surtout le sujet jeune adulte, masculin. Le tabagisme est un facteur causal. La maladie est souvent réversible à l'arrêt du tabac et facilitée par la corticothérapie.
La pneumopathie interstitielle lymphocytaire est rare, elle se rencontre surtout au cours des syndromes de Gougerot-Sjögren ou de l'infection par le VIH chez les enfants.
6
.
3
-
Les pneumopathies infiltrantes au cours des connectivites et des vascularites
Les principales maladies en cause sont la sclérodermie, la polyarthrite rhumatoïde et la myopathie idiopathique inflammatoire notamment celle associée à la présence d'auto-anticorps anti-JO-1, et le syndrome de Gougerot-Sjögren. La spondylarthrite ankylosante donne le plus souvent une fibrose dense des sommets, mutilante. La maladie de Wegener et la polyangéite microscopique donnent le plus souvent lieu à des hémorragies alvéolaires diffuses.
6
.
4
-
Les autres pneumopathies interstitielles diffuses
La lymphangioléiomyomatose est liée à une prolifération des cellules musculaires lisses. Elle touche surtout la femme en période d'activité génitale. L'aspect tomodensitométrique est caractéristique avec la présence de lésions kystiques diffuses.
Les autres pneumopathies interstitielles diffuses peuvent être considérées comme exceptionnelles.
6/7