4.
1.
8 - Manifestations musculaires (5 % à 30 % des cas)
Des myalgies ou, beaucoup plus rarement, d’authentiques myosites ont été décrites.
4.
1.
9 - Manifestations ganglionnaires et spléniques (10 % à 70 % des cas)
Des adénopathies cervicales sont fréquentes au cours d’un lupus évolutif (30 % à 70 % des cas). En cas d’examen histologique, on observe une hyperplasie folliculaire bénigne. Une splénomégalie modérée est possible, mais assez rare (10 % à 20 % des cas).
4.
1.
10 - Atteintes digestives et hépatiques
L’atteinte la plus caractéristique est la péritonite lupique mais elle est très rare (moins de 10 % des cas), parfois révélatrice.
Une atteinte hépatique (cytolyse modérée) est observée dans près de 30 % des lupus évolutifs.
Il n’y a pas d’atteinte intestinale spécifique du lupus mais des ulcérations, des perforations ou des hémorragies ont été observées, liées soit à une atteinte vasculaire (vascularite), soit au traitement (AINS, corticoïdes).
Exceptionnellement, des thromboses veineuses hépatiques ou mésentériques sont possibles dans le cadre du syndrome des antiphospholipides.
4.
1.
11 - Atteintes oculaires
Des atteintes oculaires à type de rétinite (5 % à 20 % des cas) ou d’atteinte des nerfs oculomoteurs ou du nerf optique existent.
4.
2 - Signes biologiques évocateurs
Différentes anomalies biologiques détectables dans des examens de routine peuvent orienter vers un lupus, même si elles ne sont pas spécifiques :
– une élévation de la
VS, habituellement sans élévation des protéines de l’inflammation (CRP, fibrinogène). Cette VS élevée est liée à une hypergammaglobulinémie polyclonale faite essentiellement d’IgG ;
– une cytopénie périphérique (anémie hémolytique, lymphopénie, neutropénie, thrombopénie) est très évocatrice à partir du moment où il n’y a
pas de cause toxique ou infectieuse (virale). Il existe des formes sévères (cytopénie profondes parfois combinées ; syndrome d’Evans1).
En pratique, en présence d’une cytopénie :
– il faut s’assurer de sa nature périphérique en effectuant, en cas de doute, un myélogramme ;
– il faut essayer de déterminer sa nature immunologique en recherchant des anticorps anti-globules rouges (test de Coombs) ou éventuellement d’anticorps anti-plaquettes ou anti-lymphocytes et/ou granulocytes.
Les principales manifestations cliniques et anomalies de laboratoire devant faire évoquer un syndrome des antiphospholipides sont les suivantes.
5.
1 - Manifestations cliniques évocatrices
5.
1.
1 - Thromboses vasculaires
Cf. tableau 14.II.
5.
1.
2 - Grossesses pathologiques
Cf. tableau 14.II.
5.
1.
3 - Manifestations cardiaques
Des lésions valvulaires (végétations, épaississements et dysfonctionnements) sont fréquentes au cours du syndrome des antiphospholipides, indépendamment de la coexistence d’un LED. La présence et la sévérité des lésions valvulaires doivent être documentées grâce à l’échocardiographie.
Les thromboses coronariennes s’intègrent dans le cadre plus large des thromboses vasculaires, constituant un des critères de classification du syndrome des antiphospholipides
5.
1.
4 - Manifestations cutanées
Le livedo réticulaire constitue la plus fréquente des manifestations cutanées du syndrome des antiphospholipides. Il est classiquement persistant, irréversible au réchauffement, violacé, rouge ou bleu, touchant le tronc et/ou les membres.
Les autres manifestations (ulcérations cutanées, lésions de pseudo-vascularite, gangrènes digitales, phlébites superficielles…) sont plus rares.
5.
1.
5 - Manifestations neurologiques
La spécificité de l’altération des fonctions cognitives, des céphalées ou migraines, de l’épilepsie, ou des signes de démyélinisation du système nerveux central associés au syndrome des antiphospholipides n’a pas été jugée suffisante pour les intégrer dans les critères de classification.
Les accidents vasculaires cérébraux s’intègrent dans le cadre plus large des thromboses vasculaires, constituant un des critères de classification du syndrome des antiphospholipides.
5.
1.
6 - Manifestations rénales
La présence d’anticorps antiphospholipides est corrélée à une vasculopathie rénale caractérisée par des lésions des petites artères et une ischémie rénale chronique, caractérisant la néphropathie associée aux antiphospholipides. Ces lésions sont indépendantes de l’existence d’une néphrite lupique.
5.
2 - Anomalies de laboratoire évocatrices
5.
2.
1 - Anticoagulant lupique
Cf. tableau 14.II.
5.
2.
2 - Anticorps anti-cardiolipine
Cf. tableau 14.II.
5.
2.
3 - Anticorps anti-β2-glycoprotéine I
Cf. tableau 14.II.
5.
2.
4 - Autres autoanticorps
Des anticorps anti-cardiolipine d’isotype IgA, anti-β2-glycoprotéine I d’isotype IgA, anti-phosphatidylsérine, anti-phophatidyléthanolamine et anti-prothrombine ont été décrits dans le cadre du syndrome des antiphospholipides.
5.
2.
5 - Thrombocytopénie
La thrombocytopénie est plus fréquente au cours des syndromes des antiphospholipides associés à un LED qu’au cours des syndromes des antiphospholipides isolés.
L’existence d’une thrombocytopénie est retenue si la numération plaquettaire est inférieure à 100 000 plaquettes/mm3, à au moins deux reprises, à douze semaines d’intervalle.
Cette thrombocytopénie est considérée comme étant associée au syndrome des antiphospholipides après avoir éliminé un purpura thrombotique thrombocytopénique, un syndrome hémolytique et urémique, une coagulation intravasculaire disséminée, une pseudo-thrombocytopénie et une thrombocytopénie induite par l’héparine.
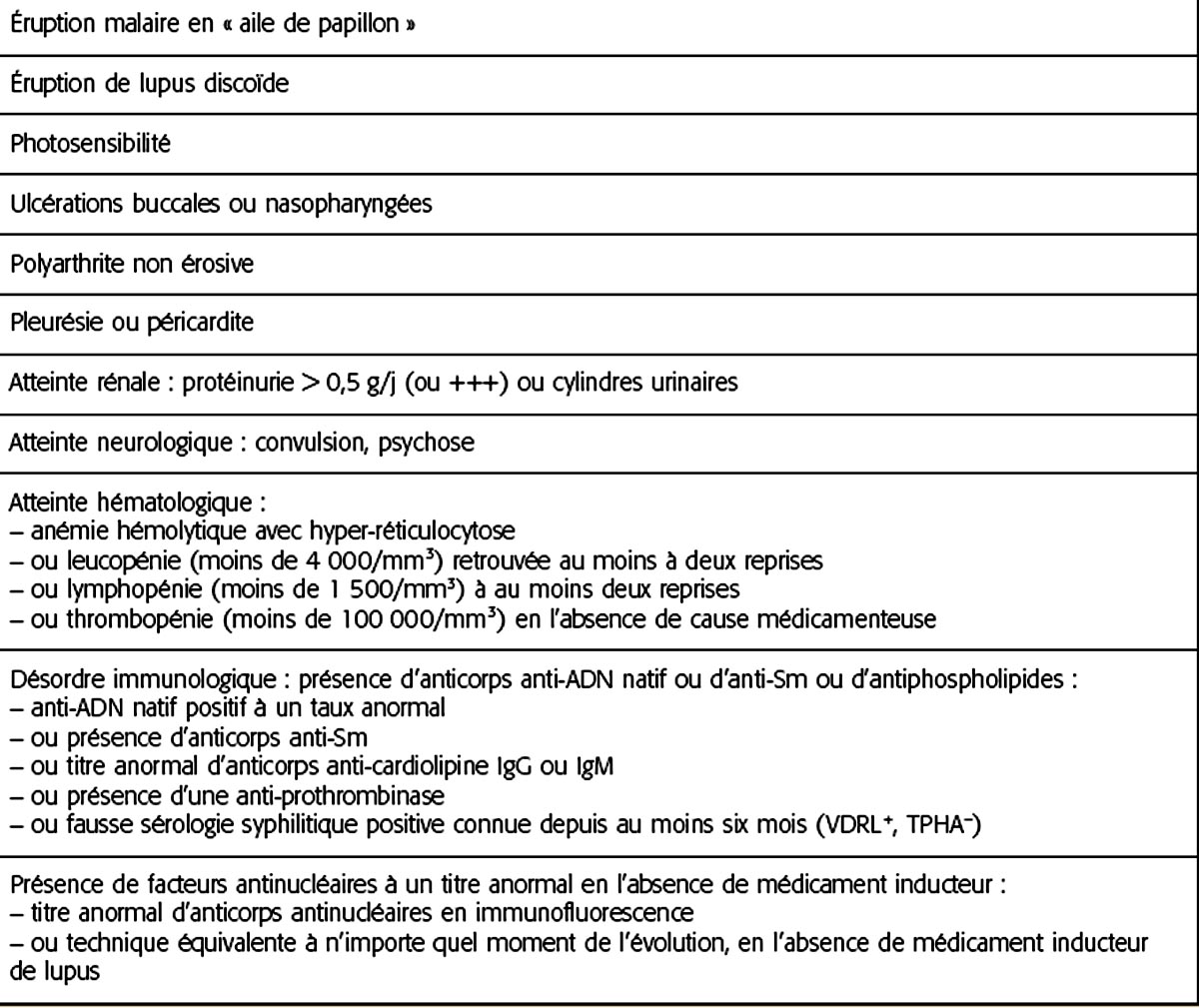
Le lupus est une affection systémique dont le diagnostic repose sur un faisceau d’arguments clinicobiologiques que résume une liste de onze critères (tableau 14.I). Cependant, ces critères ne sont pas forcément adaptés au diagnostic des formes débutantes. Ces critères intègrent en particulier la recherche d’autoanticorps qui est l’élément clé du diagnostic de lupus.
6.
1 - Recherche d’anticorps antinucléaires
Cette technique met en évidence des anticorps antinucléaires dans 99 % des lupus avec les techniques utilisées actuellement (immunofluorescence indirecte cellules Hep2). Dans un second temps, l’utilisation des techniques d’identification doit permettre de déterminer la nature de ces autoanticorps.
6.
2 - Anticorps anti-ADN natif
Ces anticorps sont très spécifiques du lupus en particulier s’ils sont à taux élevé. Néanmoins, ils ne sont pas constants, présents uniquement dans 50 % à 80 % des cas. Ils sont identifiés par des techniques immunoenzymatiques (ELISA), radioimmunologique (test de Farr) ou en immunofluorescence (Crithidia luciliae). Ils doivent être bien distingués des anticorps anti-ADN dénaturé (simple brin) qui ne sont absolument pas spécifiques.
6.
3 - Anticorps anti-antigène nucléaire soluble, ou anti-ENA
Les anticorps anti-antigène nucléaire soluble, anti-ENA (Extractable Nuclear Antigens), le plus souvent dirigés contre des ribonucléoprotéines nucléocytoplasmiques sont détectés par immunodiffusion par immuno-empreinte.
Les anticorps anti-Sm sont décrits exclusivement dans le lupus, mais ne sont présents que dans 10 % à 20 % des cas.
Les anticorps anti-Ro/SS-A et/ou anti-La/SS-B sont décrits dans 30 % à 70 % des lupus, plus particulièrement quand il existe des lésions cutanées. Ils ne sont pas spécifiques du lupus car ils sont également observés dans 40 % à 70 % des syndromes de Gougerot-Sjögren primaires.
6.
4 - Autres anticorps antinucléaires
Les anti-RNP (U1-RNP) peuvent s’observer dans 20 % à 30 % des lupus, mais ne sont pas spécifiques et sont également détectés dans les connectivites mixtes et le syndrome de Gougerot-Sjögren.
Il existe d’autres spécificités rares parfois caractéristiques du lupus (anti-PCNA), nécessitant des techniques de détection spécialisées.
6.
5 - Anticorps antiphospholipides
Ces anticorps comprennent les anticoagulants circulants, ou anti-prothrombinase, détectés par des tests d’hémostase, les anticorps anti-cardiolipine, les anticorps anti-β2-glycoprotéine-I et d’autres spécificités plus rares (anti-phosphathydiléthanolamine, anti-annexine V) détectées par des réactions immunoenzymatiques (ELISA). Ces anticorps peuvent exister isolément ou se compliquer de thromboses veineuses et/ou artérielles ou d’avortements à répétition, définissant alors le syndrome des antiphospholipides.
6.
6 - Autres autoanticorps
Des facteurs rhumatoïdes IgM sont détectés dans plus de 20 % des lupus. Les anticorps anti-histones, considérés comme spécifiques des lupus médicamenteux, sont également détectés dans 50 % à 60 % des lupus érythémateux disséminés idiopathiques.
6.
7 - Anomalies du complément total (CH50) et de ses fractions
L’hypocomplémentémie traduit généralement une consommation de l’ensemble des fractions de complément, liée à une activité de la maladie lupique. Dans ce cas, on observe une baisse du complément hémolytique total (CH50) et de l’ensemble des fractions. En pratique, ce sont généralement les fractions C3 et C4 qui sont analysées par méthode immunochimique.
La baisse isolée d’une fraction (C2, C4, C1q), généralement sans baisse du complément hémolytique total, est l’expression d’un déficit congénital qui est une anomalie génétique assez fréquente au cours du lupus. Il ne faut pas confondre cette anomalie avec une hypocomplémentémie de consommation.
Au total, l’étude du complément n’est pas utile au diagnostic, mais peut servir pour le suivi évolutif d’un lupus.
En pratique, le diagnostic de lupus repose donc sur l’analyse de tous ces éléments cliniques et biologiques (figure 14.1). Le problème le plus fréquent est celui d’un lupus débutant. Dans ce cas, soit les signes observés sont très spécifiques (érythème lupique, anticorps anti-ADN natif), permettant de retenir le diagnostic, soit une surveillance prolongée permettra d’affiner le diagnostic.
Comme dans le cadre du LED, le diagnostic de syndrome des antiphospholipides repose sur un faisceau d’arguments cliniques et de laboratoire, dont les plus spécifiques ont servi de base à l’élaboration de critères de classification pour le syndrome des antiphospholipides, révisés en 2006 (tableau 14.II).
Bien qu’il ne s’agisse que de critères de classification, ces critères cliniques et de laboratoire sont fréquemment utilisés pour retenir un diagnostic de syndrome des antiphospholipides.
Le comité ayant révisé ces critères de classification pour le syndrome des antiphospholipides déconseille l’utilisation du terme de syndrome « secondaire » des antiphospholipides, puisque la traduction clinique des antiphospholipides est comparable qu’il s’agisse d’un syndrome dit « primaire » ou dit « secondaire » des antiphospholipides (associé à un LED par exemple). En revanche, il est recommandé de documenter la coexistence d’un LED ou d’une autre maladie auto-immune associée au syndrome des antiphospholipides, en raison notamment des implications thérapeutiques de ces maladies auto-immunes associées. Enfin, nous ne ferons que citer ici le syndrome catastrophique des antiphospholipides qui associe, simultanément ou en moins d’une semaine, des manifestations cliniques traduisant une atteinte multiviscérale (atteinte d’au moins trois organes, systèmes ou tissus), traduisant des occlusions vasculaires confirmées par un examen anatomopathologique, chez un individu présentant des anomalies de laboratoire caractéristiques du syndrome des antiphospholipides (anticoagulant lupique ou anticorps anti-cardiolipine).
7/14