4 - Diagnostic de l’hémochromatose
La découverte du gène HFE a profondément modifié la stratégie diagnostique. Il faut cependant se rappeler que la mutation C282Y à l’état homozygote n’est pas suffisante pour produire la maladie.
On doit différencier deux situations :
- la suspicion d’hémochromatose devant des manifestations cliniques ou paracliniques ;
- le dépistage familial chez un patient asymptomatique, apparenté au 1er degré à un sujet génétiquement atteint.
4.
1 - Suspicion d’hémochromatose
Le diagnostic est souvent évoqué devant :
- des manifestations cliniques peu spécifiques : asthénie, perte de poids, douleurs articulaires, douleurs abdominales, diminution de la libido, myocardiopathie atypique ;
- des manifestations biologiques : élévation des marqueurs sériques du fer, élévation des transaminases, hyperglycémie ou diabète « de type 2 » ;
- des anomalies morphologiques : hépatomégalie, ostéoporose.
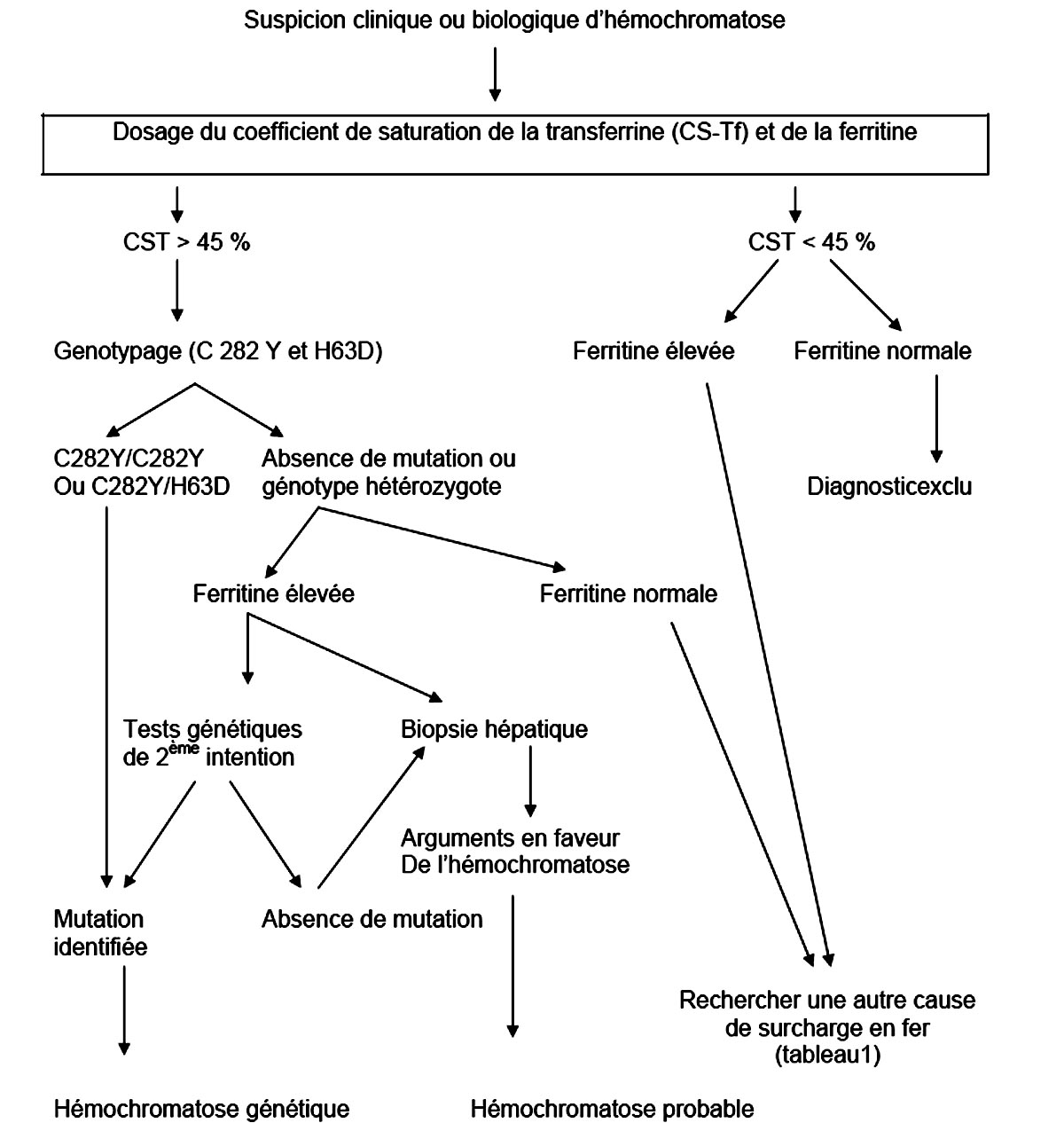
La stratégie diagnostique est schématisée sur la figure 15.2.
1. Première étape
La première étape consiste à affirmer biochimiquement l’anomalie du métabolisme du fer. Il faut mesurer le coefficient de saturation de la transferrine (CS-Tf), qui constitue le marqueur le plus sensible et spécifique de la maladie (+++). Pour un seuil fixé à 45 %, sa sensibilité pour dépister une hémochromatose primitive est de 81 % chez l’homme et de 48 % chez la femme, et sa spécificité est de 94 % et 97 %, respectivement.
Le dosage de la concentration plasmatique de la ferritine permet d’estimer les réserves en fer de l’organisme. Une valeur supérieure à 300 mg/L chez l’homme et à 200 mg/L chez la femme est en faveur d’une surcharge en fer, mais de fausses augmentations sont observées au cours des pathologies inflammatoires. Par ailleurs, au cours de l’hémochromatose, la ferritinémie n’augmente que tardivement lorsque survient la surcharge hépatique en fer. Son dosage permet d’apprécier le stade évolutif de la maladie (+++). Le risque de cirrhose serait nul tant que la ferritinémie est inférieure à 1 000 mg/L et que les transaminases sont normales.
a. CS-Tf supérieur à 45 %
À cette condition, la suspicion d’hémochromatose primitive est forte. L’élévation de la ferritinémie renforce cette présomption mais sa normalité n’élimine pas le diagnostic (+++).
b. CS-Tf inférieur à 45 %
À cette condition, l’hémochromatose primitive peut être éliminée. La constatation fréquente d’une ferritinémie élevée associée à un CS-Tf bas doit faire évoquer une autre cause de surcharge en fer (cf. tableau 15.I), et en particulier :
- l’hépatosidérose dysmétabolique ou NASH (non alcoholic steatosis hepatitis), observée au cours des syndromes d’insulinorésistance, associant une élévation des ALAT, des gGT et de la ferritinémie à un foie stéatosique à l’échographie. Il s’agit d’une cause fréquente de surcharge en fer, susceptible d’évolution vers la fibrose hépatique, qui tire bénéfice de la prise en charge de l’insulinorésistance et éventuellement d’une déplétion martiale ;
- l’acéruléoplasminémie héréditaire, qui peut s’accompagner d’un diabète et d’une surcharge hépatique en fer, avec des signes neurologiques (syndrome extrapyramidal, ataxie, démence) n’existant pas dans l’hémochromatose. Il s’agit d’une affection rare liée à un déficit de l’activité céruloplasmine ferroxidase (la céruloplasminémie est indétectable) ;
- la surcharge hépatique en fer par mutation du gène de la ferroportine 1. Il s’agit d’une maladie exceptionnelle, décrite récemment, de transmission dominante avec surcharge en fer macrophagique. La réponse au traitement par déplétion martiale est faible ou nulle (cf. tableau 15.II).
2. Deuxième étape
La deuxième étape consiste à rechercher une mutation C282Y ou H63D du gène HFE, par analyse génétique, après consentement écrit du patient.
a. Patient homozygote C282Y ou hétérozygote composite
Si le patient est homozygote C282Y +/+ ou hétérozygote composite C282Y/H63D, le diagnostic d’hémochromatose est acquis (HFE1). La ferritinémie permet d’estimer la surcharge ferrique et d’orienter la prise en charge (tableau 15.IV).
Lorsque la ferritinémie est élevée, il existe un risque de retentissement viscéral et métabolique :
- il est indispensable de pratiquer des examens complémentaires : glycémie à jeun, dosage des transaminases, échographie abdominale, ECG et, en fonction du contexte clinique, radiographies articulaires, échographie cardiaque, bilan hormonal avec dosage de testostérone chez l’homme, ostéodensitométrie osseuse s’il existe des cofacteurs d’ostéoporose ;
- en fonction des résultats, il sera nécessaire de confier le patient à un spécialiste. Le recours au spécialiste est systématique si la ferritine est supérieure à 1 000 mg/L, pour discuter la réalisation d’examens complémentaires (mesure de la surcharge hépatique en fer par IRM quantitative). La ponction-biopsie hépatique n’est pratiquée qu’en cas de suspicion de fibrose et pour rechercher des signes de gravité (cirrhose, cancer hépatocellulaire).
b. Patient hétérozygote simplex ou absence de mutation
Si la recherche de mutation est négative ou si le patient est hétérozygote simplex pour C282Y ou H63D, il faut être très critique vis-à-vis du diagnostic d’hémochromatose.
Il faut se souvenir qu’une élévation du CS-Tf n’est pas totalement spécifique de l’hémochromatose (tableau 15.V). Il faut donc toujours confronter les données clinicobiologiques.
Si le contexte est très évocateur et qu’il existe une élévation persistante de la ferritinémie, on évoquera une autre forme d’hémochromatose héréditaire (hémochromatose juvénile, mutation du gène du récepteur de la transferrine, cf. tableau 15.II), à rechercher par des tests génétiques de 2eintention. En pareil cas, on proposera le plus souvent une ponction-biopsie hépatique qui pourra donner des arguments en faveur d’une probable hémochromatose héréditaire (index de surcharge ferrique, distribution hépatocytaire du fer).
La biopsie hépatique, autrefois couramment utilisée pour le diagnostic, n’est donc utilisée actuellement que :
- soit à visée diagnostique, en cas d’anomalie ferrique avec enquête génétique négative ;
- soit à visée pronostique, en cas de suspicion d’atteinte hépatique sévère.
4.
2 - Dépistage familial
Le dépistage est proposé de manière systématique chez les apparentés du premier degré. Compte tenu du caractère généralement tardif des manifestations cliniques, il est réalisé chez le jeune adulte. Un bilan martial perturbé conduit d’emblée à la réalisation d’un test génétique pour confirmer le diagnostic.
La normalité du bilan martial n’exclut pas le diagnostic. On peut proposer :
- une surveillance régulière du bilan martial ;
- un dépistage génétique qui, dans le cadre de la loi sur le dépistage des apparentés sains des patients porteurs de maladie génétique, sera réalisé avec l’aide du patient, chargé d’informer les apparentés et après accord de ces derniers, par un généticien ou un spécialiste œuvrant dans le cadre d’un réseau spécialisé. Le dépistage permettra d’interrompre toute surveillance chez les sujets indemnes de la mutation et leurs descendants. Chez les sujets atteints, le diagnostic sera ainsi posé à un stade totalement asymptomatique et conduira à la réalisation d’un bilan martial tous les 3 ans, en se souvenant que tous les sujets porteurs de la mutation ne présenteront pas la maladie (cf. tableau 15.IV).


4/8