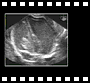
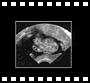
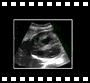
 Voie abdominale
Voie abdominale Voie abdominale
| 5 - Gyration |
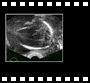
5.2. Pachygyre-lissencéphalie :
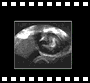
| 6 - Tube neural |
Dysraphie médullaire :
Ce sont des malformations de la moëlle, de ses enveloppes ou du canal rachidien, consécutives à une anomalie de fermeture du tube neural. Les malformations observées sont diverses avec ou sans traduction clinique.a) Spina Bifida
Il correspond à un défaut de fermeture osseux. La fréquence varie selon le pays; en France 5/10 000 naissances. Le risque de récurrence est de 5%. Le contenu de la poche et sa situation définissent plusieurs formes cliniques :b) Myélodysraphie occulte
Ce sont des anomalies médullaires habituellement basses (L5-S1) sans hernie du tissu nerveux et recouvrement cutané normal. Ils sont souvent « parlant » cliniquement et ont le même risque de récurrence que le spina bifida. Ce sont : Filum Terminal court, Fistules dermiques, Syringomyélie, Syndrome de régression caudale.c) Malformations de la charnière occipito-cervicale
Elles peuvent concerner soit l'os occipital, soit les vertèbres cervicales avec des anomalies de nombre, de forme ou de jonction.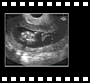 13 semaines
13 semaines

6.3 Myélo-méningocèle :
 |
Voie abdominale |  |
|
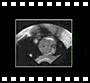 |
Précoce vaginale | 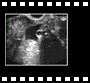 |
Petite taille vaginale |
| 7 - Rappel sur les pathologies malformatives |
7.1 Crâniosténoses :
Elles sont dues à une soudure prématurée d'une ou de plusieurs sutures,elles provoquent une déformation de la forme du crâne avec atteinte cérébrale et occulaire possible. Certaines peuvent être d'expression anté-natale :- l'acrocéphalo-syndactylie ou syndrome d'APERT se manifeste par une proéminence antérieure du crâne et une syndactylie,
- la dysostose crânio-faciale ou maladie de CROUZON se caractérise par une acrocéphalie et un hypertélorisme. Elle se transmet sur le mode autosomique dominant,
- le crâne en trèfle, parfois associé à d'autres malformations en particulier digestives, parfois un nanisme,
- la scaphocéphalie : la forme du crâne est allongée et rétrécie. L'anomalie touche les garçons.7.2 Les ostéochondrodysplasies :
2 types d'ostéochondrodysplasies sont accessibles au diagnostic anténatal échographique : celles qui s'accompagnent d'anomalies de croissance et celles qui s'accompagnent d'anomalies de la densité osseuse.7.2.1 Ostéochondrodysplasie avec anomalie de la croissance :
Parmi ces chondrodysplasies, certaines sont toujours létales et d'autres peuvent être compatibles avec la vie.7.2.1.1 Les chondrodysplasies toujours létales :
a) Le nanisme tanatophore
Fréquence 1/6400 naissances selon Doumenfeld et 1/100.000 selon Lang.- nanisme micromélique,
- un thorax étroit,
- un abdomen proéminent,
- une macrocéphalie réalisant parfois un aspect de crâne en tréfle.
b) L'achondrogénèse
C'est la chondrodysplasie létale la plus sévère. Elle associe un nanisme micrométique extrême avec macrocéphalie et brièveté du tronc.- l'achondrogénèse de type I : la forme la plus rare où les os longs sont réduits à des amas calcifiés plus ou moins informes et souvent méconnaissables.
- l'achondrogénèse de type II où le nanisme est moins sévère et le défaut d'ossification des os longs est moins marqués.
Les deux signes échographiques principaux de l'affection sont la macrocéphalie et la micromélie. Un hydramnios et un anasarque foetal avec infiltration des téguments prédominant dans la région cervicale sont fréquents.c) Le syndrome polydactylie - Côtes courtes
Il représente un troisième groupe de chondrodysplasie létale dont la transmission génétique se fait sur un mode récessif antosmique (25% de risque de récurrence).- Saldino-Noonan ou
type I,
- Majewski ou type II,
- Nanmoff ou type III.
- la micromélie à prédominance
acromélique,
- la polydactylie et l'étroitesse du thorax.
- sténose digestive,
- hypoplasie rénale,
- polykystose rénale,
- oreillette et ventricule unique,
- communication inter-auriculaire,
- transposition des gros vaisseaux,
- hydrocéphalie,
- micrognathisme,
- et hydramnios ou oligoamnios selon les malformations viscérales associées.
7.2.1.2 Les chondrodysplasies compatibles avec la vie :
Elles peuvent être réparties en trois groupes selon que la micromélie est l'élément essentiel ou bien qu'elle l'accompagne d'un racourcissement du tronc ou d'autres anomalies.a) Les chondrodysplasies avec micromélie prépondérante
- l'achondroplasie
Sa fréquence varie de 1 à 25000 à 1 pour 66000 naissances. La transmission est autosomique dominante. Les mutations récentes sont fréquentes, de l'ordre de 85%, parfois favorisées par l'élévation de l'âge paternel. Dans la forme homozygote, le nanisme est précoce et sévère. L'évolution est en règle générale fatale dans les premiers jours ou les premières semaines de la vie. Dans le forme hétérozygote le pronostic est habituellement favorable en dehors de l'insuffisance staturale. La taille ne dépasse pas 1m25. L'intelligence est normale.- le nanisme diastrophique
Cette affection se caractérise à la naissance par un nanisme micromélique à prédominance rhizomélique et acromélique associé à des anomalies des extêmités et de la face. La transmission se fait selon un mode récessif autosomique. Létale dans 25% des cas au cours de la petite enfance. Chez le grand enfant, le nanisme est sévère . L'intelligence est normale.- la chondrodysplasie ponctuée
Caractérisée par la présence de foyers de calicifications au voisinage des cartilages épiphysaires et des vertèbres. La fréquence st de 0,09 pour 10000 naissances selon Romero. Le mode de transmission est hétérogène : une transmission sur le mode dominant liée à l'x est habituelle; néanmoins, une transmission sur le mode récessif autosomique est possible.- les dysplasies mésoméliques
Caractérisée par une extrême micromélie qui intéresse électivement l'avant-bras et la jambe. La transmission est récessive autosomique.- la dysplasie métatropique
Se caractérise par l'aspect allongé et étroit du thorax contrastant avec la brièveté des membres; le crâne reste de taille normale. L'atteinte osseuse touche électivement le rachis, le bassin et les membres. La transmission pourrait être récessive autosomique.- la dysplasie acromésomélique
Pourra être évoquée in utero devant la brièveté de l'avant-bras, de la jambe et de l'aspect trapu des mains et des pieds. La transmission est récessive autosomique.b) Chondrodysplasie avec insuffisance staturale globale
- La maladie de Kwieff,
- la dysplasie spondylo-épiphysaire congénitale.
c) Chondrodysplasie avec retard statural et anomalies associées
- la dysplasie chondro-ectodermique
Qui associe une micromélie de type mésomélique, une polydactylie postaxiale des doigts et parfois des orteils, des anomalies ectodermiques qui intéressent les ongles et les dents, un thorax long et étroit et une cardiopathie congénitale dans 50% des cas. La transmission est autosomique récessive.- la dysplasie thoracique asphyxiante
Cette chondrodysplasie autosomique récessive sera évoquée devant toute nanisme micromélique modéré à thorax très étroit et côtes courtes.- la dysplasie cléido-crânienne
Défaut d'ossification de la voûte du crâne et des anomalies claviculaires à type d'agénésie ou d'aplasie. Les troubles de la croissance sont modérés. Le thorax est parfois étroit. La transmission se fait sur un mode dominant.7.2.2 Ostéochondrodysplasie avec anomalie de la densité osseuse :
7.2.2.1 Ostéochondrodysplasie avec transparence excessive du squelette :
a) Ostéogénèse imparfaite :
Maladie
du collagène, héréditaire, caractérisée par une ostéoporose vraie, des sclérotiques
bleues, une surdité progressive, une dentinogénèse imparfaite, une hyperlaxité
ligamentaire et des anomalies de certains métabolismes, notamment de la fonction
plaquettaire. C'est une maladie rare dont le taux de survenue est de 1/20.000
à 1/60.000 naissances. On regroupe sous le terme un ensemble d'affections différentes
par leur aspect clinique et leur mode de transmission.
Différents
types de l'ostéogénèse imparfaite selon Sillence (1979) :
TYPE |
SYNONYME |
TRANSMISSION |
Signes
cliniques et radiologiques |
Létale
à la naissance |
I |
Syndrome de Van der Hoeve Osteogenesis imperfecta tarda levis Syndrome de Eddowe | D |
Nanisme modéré Les fractures surviennent après la naissance, parfois au cours du 3ème trimestre Sclérotiques bleues | non |
II |
Ostéogénèse imparfaite congénitale létale Ostéogénèse imparfaite de Vrolik | r |
Nanisme sévère Fractures et cals osseux nombreux Sclérotiques Bleues Déformations majeures des membres | oui |
III |
Ostéogénèse imparfaite tarda gravis Ostéogénèse imparfaite congénitale type Vrolik Dysplasie périostale de Porak et Durante Ostéopsathyrose de Lobstein | r |
Déformation progressive et sévère des membres et de la colonne vertébrale post-natale Les sclérotiques sont bleues (le bleu s'atténue avec l'âge) | non |
IV |
Maladie de Ekman-Lobstein Ostéopsathyrose idiopathique de Lobstein | D |
Taille normale Sclérotiques blanches Manifestation atténuée de la maladie | non |
b) Hypophosphatésie :
Maladie osseuse secondaire à un désordre du métabolisme phosphocalcique d'origine génique ayant pour conséquence une transparence excessive des os.Les principaux signes échographiques de l'affection : le pôle céphalique qui est à peine visible et déformable, les os longs sont courts et incurvés et possèdent une faible échogénicité.
7.2.2.2 Ostéochondrodysplasie avec hyperdensité osseuse :
- Maladie de Coffey
Elle touche électivement la diaphyse des os longs dont la corticale l'épaissit en fuseau et se densifie secondairement.
7.3 Anencéphalie :
Anomalie
globale, définie par l'absence de fermeture de tube neural à son extrêmité céphalique.
Elle se diagnostique précocément dès 13 SA.
Elle se caractérise par :
- l'absence de voûte
crânienne,
- un hydramnios fréquent,
- des mouvements souvent nombreux, saccadés et brusques.
Elles sont associées dans 50% des cas à un rachichisis. Dans 30% des cas à des malformations viscérales ou à des anomalies faciales.
Après le 5ème mois, la survenue d'un hydramnios constitue un signe d'appel classique.7.4 Encéphalocèle :
Extériorisation du tissu cérébral à travers une brèche crânienne. Leur fréquence est de 0,5 pour 1000 naissances.7.5 Anévrisme de l'ampoule de Galien :
Il est caractérisé par l'association de :- l'image liquidienne en raquette correspondant à la dilatation kystique de l'ampoule, médiane ou légèrement excentrée prolongée par le sinus droit dilaté,
- l'hyperpulsatilité artérielle des vaisseaux du cou et du polygone de Willis et la turgescence des jugulaires,
- l'asystolie traduite par une cardiomégalie.
7.6 Syndrome d'Aicardi :
Associe agénésie du corps calleux, dilatation ventriculaire avec hétéropie périventriculaire de la substance grise, chez un foetus du sexe féminin, avec anomalie de la segmentation rachidienne.7.7 Syndrome de Dandy-Walker :
Associe une dilatation kystique du 4ème ventricule à une agénésie ou une hypoplasie du vermis ; il peut coexister une agénésie du corps calleux et une hydrocéphalie d'importance variable.7.8 Holoprosencéphalie :
Anomalie du clivage du cerveau antérieur, l'holoprosencéphalie regroupe un ensemble de malformations siègeant au niveau de l'encéphale et de la face. L'aspect anatomique est variable selon le degré de division de prosencéphale.7.9 Hydranencéphalie :
C'est une structure liquidienne anormale. Le manteau cérébral est réduit à une fine membrane. Le faux du cerveau est réduite voire absente. Une vaste plage liquidienne, unique, surmonte l'image des thalami non fusionnés et du cervelet.7.10 Kystes arachnoïdiens :
Correspondant à une anomalie du développement de l'arachnoïde avec dédoublement de sa membrane. Ils ont une topographie préférentielle au niveau de la vallée sylvienne, de l'angle ponto-cérébelleux, de la plaque quadrigéminale, de la citerne cérébelleuse, de la scissure interhémisphérique et du sillon inter-pédonculaire.7.11 Agénésie du corps calleux :
Elles peuvent être totales ou partielles, isolées ou associées.- non identification
du septum lucidum,
- absence ou interruption du faux du cerveau,
- isolée ou associée à une hydrocéphalie,
- elle doit être recherchée systématiquement en cas de malformations faciales
ou d'anomalies vertébrales (hemivertèbres).
7.12 Malformations d'Arnold-Chiari type II :
Correspond à une ectopie de la partie inférieure du vermis, de la protubérance et du 4ème ventricule dans le trou occipital. La fosse postérieure est étroite ; les hémisphères cérebelleux sont hypoplasiques; une dilatation ventriculaire est fréquemment associée. Elle est retrouvée dans 65% à 100% des formes graves de spina-bifida. La reconnaissance directe est assez difficile à l'échographie surtout après 23 SA. En revanche elle peut être indirectement prouvées par les anomalies positionnelles de l'encéphale et du cervelet qu'elle entraîne.7.13 Dysraphie médullaire :
Ce sont des malformations de la moëlle, de ses enveloppes ou du canal rachidien, consécutives à une anomalie de fermeture du tube neural.7.13.1 Spina Bifida :
Il correspond à un défaut de fermeture osseux.- méningocèle : hernie isolée des méninges,
- myéloméningocèle : hernie comportant les méninges et les tissus nerveux responsable de paraplégie
plus ou moins étendue,
- spinalipome : c'est une tumeur lipomateuse associée à un défect
cérébral. L'adhérence moëlle-lipome est étroite.
On distingue les localisations lombo-sacrées (50-60%) ou dorsales basses (30%) et les localisations dorsales hautes ou cervicales qui s'accompagnent d'une paraplégie spastique de mauvais pronostic. De plus, 95% des enfants présentant un spina bifida lombaire ou lombo-sacrée ont également des anomalies au niveau de la moëlle cervicale ou une malformation d'Arnold-Chiari type II à l'origine d'une hydrocéphalie.
7.13.2 Myélodysraphie occulte : Ce sont des anomalies médullaires habituellement basses (L5-S1) sans hernie du tissu nerveux et recouvrement cutané normal. Ils sont souvent « parlant » cliniquement et ont le même risque de récurrence que le spina bifida. Ce sont :
- Filum Terminal court,
- Fistules dermiques,
- Syringomyélie,
- Syndrome de régression caudale.
7.13.3 Malformations de la charnière occipito-cervicale :
Elles peuvent concerner soit l'os occipital, soit les vertèbres cervicales avec des anomalies de nombre, de forme ou de jonction.