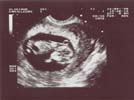

1 - Echographie normale
2 - Aspects pathologiques
3 - Agénésies et Hypoplasies
4 - Brièveté des membres
5 - Anomalies de segmentation
6 - Malposition des membres
7 - Syndrome d’Immobilisme fœtal
8 - Tératogénicité et anomalies de membres
9 - Conclusion
Introduction
:
Les appareils actuels en temps réel de haute résolution permettent
une étude morphologique et biométrique très précoce
des membres et des extrémités fœtales. Des courbes de croissances
des différents segments de membres ont été réalisées
dans le but d’améliorer le dépistage anténatal
précoce des malformations squelettiques qu’il s’agisse
des chondrodysplasies ou des agénésie partielles ou totales
des membres.
L’échographie morphologique des membres fœtaux est un examen
difficile.
Sensibilité du dépistage :
• Anomalies majeures = 70 %
• Anomalies « mineures » = 18 %
Le diagnostic anténatal des anomalies de membres doit être systématique et la période optimale est de 12 à 26 SA.
• 8 SA = visualisation
des membres
• 10 SA = pieds / mains (pouce en abduction visible)
• 12 à 14 SA = détails des doigts et orteils (on parle
de « rayons »)
La morphogenèse des membres se déroule entre la 7ème et la 10ème semaine d’aménorrhée. Le membre supérieur est constamment en avance de quelques jours sur le membre inférieur.
L’étude morphologique n’est pas concevable sans l’étude de la motilité.
1) Etude globale des
membres
* Symétrie des deux membres supérieurs puis inférieurs
associés aux mouvements de flexion-extension
* Motilité
* Aspect
2) Etude précise
des différents segments
* Biométrie
* Morphologie
* Motilité
En complément des données échographiques, certains examens seront pratiqués (radiographie du contenu utérin, prélèvements fœtaux.
Morphologie normale des membres
Les membres sont visibles à partir de la 10ème semaine sous forme de petits échos linéaires,
mais leur segmentation n’est correctement objectivée qu’à partir de la 13-14ème semaine.
A partir de la hache fœtale
facilement visualisée en coupe transversale. Les éléments
osseux de la ceinture pelvienne (os iliaque tête fémorale et
ischion forment un triangle osseux qui jouxte l’image anéchogène
de la vessie. La rotation de la sonde autour de l’articulation ilio-fémorale ![]() permet de visualiser le fémur, structure très échogène
d’aspect linéaire avant la 22èmesemaine
permet de visualiser le fémur, structure très échogène
d’aspect linéaire avant la 22èmesemaine ![]() , et d’aspect en « canne de golf » après
, et d’aspect en « canne de golf » après ![]()
![]() . Pour le mesurer, il faut obtenir sur le même axe la diaphyse dans
sa plus grande longueur ainsi que les articulations sus et sous jacentes.
Les calibreurs doivent être placés au niveau des extrémités
osseuses.
. Pour le mesurer, il faut obtenir sur le même axe la diaphyse dans
sa plus grande longueur ainsi que les articulations sus et sous jacentes.
Les calibreurs doivent être placés au niveau des extrémités
osseuses.
L’axe tibio-fibulaire est repéré par rotation de la sonde autour du genou. Ces deux os sont très rarement visibles simultanément dans leur intégrité. La fibula, latérale est grêle et linéaire ; le tibia est médial, plus échogène, épais et de forme linéaire ou en canne de golf.
L’étude des pieds nécessite deux incidences et comporte une étude statique et dynamique.
Incidence de profil :
- Etude de l’extrémité inférieure de la jambe
- Talon et avant pied
- Articulation de la cheville, mouvement de flexion-extension
Incidence de face :

- Face plantaire (éventail des orteils large, partie plus étroite
et arrondie du côté du talon)
- Les métatarses sont ossifiés vers le 4ème mois, le
calcanéum entre le 5 et le 6ème mois.
Une coupe dans l’axe de la jambe recherchera une malposition du pied.
Etude des membres supérieurs
A partir de l’épaule fœtale. Les éléments de la ceinture scapulaire (omoplate, clavicule, tête humérale) forment un triangle osseux proche du gril costal. L’omoplate est reconnue en coupe tangentielle ; pour visualiser les clavicules, il faut une coupe transversale, légèrement oblique, proche de la base du cou. L’axe huméral est déterminé par rotation de la sonde autour de l’articulation scapulo-humérale. L’humérus est très échogène et linéaire.
Os de l’avant-bras
: Retrouvé par rotation de la sonde autour de l’articulation
du coude dès la 14ème SA.
Ulna identifié à partir du coude dans le prolongement de l’olécrane.
Radius identifié à partir du poignet, son étude est importante
dans certains syndromes malformatifs avec hypoplasie ou agénésie
radiale.
L’étude de la main est statique et dynamique.



Mensurations normales des membres
Ces mesures exigent une
grande rigueur, les mesures doivent être prises dans le plus grand axe
de la diaphyse mesurée. Pour chacun des segments de membres, il existe
des courbes de croissance normale qui doivent servir de référence.
Le fémur est l’os le plus facilement mesurable et sa mesure est
la plus reproductible. L’homogénéité de l’ombre
acoustique est un critère de bonne mesure.
En pratique, la mesure de tous les segments de membres ne se fera que lorsque
la mesure du fémur sera discordante avec l’âge gestationnel.
Maturation osseuse
Elle est appréciée
sur l’apparition et la taille des points d’ossification échographique
du genou.
Point d’ossification fémoral inférieur (point de Béclard)
:
Image échogène au pôle inférieur du fémur
en dedans du genou.
Apparaît dès la 32ème SA en échographie, est toujours
présent à la 35ème SA dans les grossesses normales. Sa
taille varie de 2 mm à 33 SA à 8-10 mm à terme.
Point d’ossification
tibial supérieur (point de Todt) :
Apparaît en échographie vers la 36-37ème SA sous la forme
d’un écho dense surplombant la crosse de la « canne de
golf » tibiale.
Ainsi, au-delà de la 37ème SA, les échos des deux points
d’ossification épiphysaire du genou peuvent être visualisés.
Les circonstances qui amènent
au dépistage anténatal des anomalies des membres sont de trois
ordres :
- Antécédents familiaux d’ostéo-chondrodysplasie.
- Anomalie de la grossesse (RCIU, hydramnios, oligoamnios, diminution des mouvements
actifs fœtaux).
- Découverte fortuite au cours de l’échographie morphologique.
Le taux de détection global des agénésies est de l’ordre de 11,5 %.
Agénésies transverses
Formes terminales ou distales :
Volontiers d’origine génétique lorsqu’elles sont bilatérales et sont plus souvent associées à d’autres pathologies quand elles touchent les membres inférieurs plutôt que les membres supérieurs.
Amélie
![]() : Peut porter sur un ou plusieurs membres. Unique, elle a le plus souvent
un caractère isolé, quadruple elle est plutôt d’origine
médicamenteuse ou infectieuse.
: Peut porter sur un ou plusieurs membres. Unique, elle a le plus souvent
un caractère isolé, quadruple elle est plutôt d’origine
médicamenteuse ou infectieuse.
La maladie des brides amniotiques :

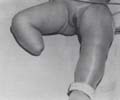

-Œdème des membres
atteints
-Présence d’un anneau de striction
-Intérêt du Doppler couleur pour évaluer le degré
de striction (diminution du flux vasculaire)
Hémimélie partielle ou totale : Peut appartenir à des syndromes comme l’aglossie-adactylie (rétrognathie, aglossie, hydramnios, amputation de membres).


Agénésie
des extrémités :
- des mains, habituellement isolée
- des pieds ![]()
![]() (achéiropodie), parfois syndrome récessif autosomique
(achéiropodie), parfois syndrome récessif autosomique
Adactylie
:
- d’une grande variété et d’un grand polymorphisme
![]()
![]()
![]()
- isolées ou entrant dans des syndromes divers incluant essentiellement
une pathologie buccale
- existent des formes familiales de caractère autosomique dominant
Ectrodactylie : Atteinte de la partie moyenne de la main ou du pied
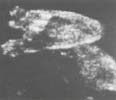
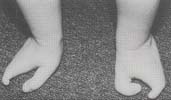
Les
phocomélies, agénésies transverses d’un segment moyen
ou proximal
Préservent la main et le pied.
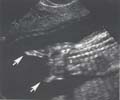

Rôle de la thalidomide dans les phocomélies complètes.
Syndrome de Roberts :
- RCIU sévère et constant
- anomalies cranio-faciales à type de fente labio-palatine et de microcéphalie
- malformations des membres allant de la phocomélie partielle à
la tétraphocomélie
- retard mental constant
- transmission autosomique récessive
Agénésies longitudinales
Soit radiale, soit ulnaire
Soit tibiale, soit fibulaire
Sont associés à des anomalies de position des extrémités.
Hypoplasies
Longitudinales, elles peuvent recouvrir différents syndromes.
CHILD syndrome (Congenital
Hemidysplasia Ichtyosis Limb Defect):
- Atteinte unilatérale des extrémités (de la simple hypoplasie
des phalanges à l’absence d’un membre
- Alopécie, atteinte cutanée ipsilatérale
- Erythème, hyperkératose
- Hydronéphrose, agénésie rénale ou cardiopathie
- 19 filles pour 1 garçon
Syndrome
de régression caudale :
De l’agénésie lombo-sacrée à la sirénomélie
![]() (malformation atteignant strictement les membres inférieurs, et incluant
des anomalies viscérales (agénésie rénale, vésicale,
rectale avec imperforation anale). Etiologies multiples (complications vasculaires,
diabétiques…)
(malformation atteignant strictement les membres inférieurs, et incluant
des anomalies viscérales (agénésie rénale, vésicale,
rectale avec imperforation anale). Etiologies multiples (complications vasculaires,
diabétiques…)
Hypoplasie
fémorale :
- Totale ou partielle ![]()
- Associé à un faciès particulier (petit nez avec hypoplasie
des ailes du nez, rétrognathisme, fente palatine)
- La majorité des cas est sporadique.
Différentes catégories
sous cette dénomination :
- Atteinte bilatérale portant sur un ou plusieurs segments de membres,
symétriquement, à la fois au niveau des membres inférieurs
et supérieurs (ex : chondrodysplasie)
- Atteinte bilatérale portant sur un segment de membre (ex : anomalies
chromosomiques)
- Atteinte touchant l’un ou l’autre segment de membre de façon
anarchique réalisant des asymétries de croissance (ex : syndrome
de Klippel-Trenaunay)
Dysplasies du squelette ou maladies osseuse constitutionnelles
Cinq groupes :
1. Ostéochondrodysplasies
:
- Anomalies de taille (nanisme) et/ou de forme des os et/ou cartilages
- Prévalence de 2,3 à 4,7 pour 10 000 naissances
2. Dysostoses :
Malformations d’un os isolé parfois associée à d’autres
malformations
3. Ostéolyses idiopathiques
4. Anomalies chromosomiques
:
Les malformations du squelette et des membres sont le signe d’appel d’anomalies
chromosomiques dans 7 % des cas environ (surtout trisomie 13 et 18).
- Pied bot (en particulier bilatéral) : évoque essentiellement
une trisomie 18
- Main bote : évoque une trisomie 13 ou 18
- Main crispée : évoque une trisomie 18
- Fémur court (<P5, rapport Fémur/Pied < 0,84) : évoque
une trisomie 21
5. Affections métaboliques
primitives
Les ostéochondrodysplasies
et les anomalies chromosomiques composent l’essentiel du dépistage
anténatal.
Classification :
- Nanisme rhisomélique : Atteinte proximale
- Nanisme mésomélique : Atteinte du segment moyen
- Nanisme acromélique : Atteinte du segment distal (mains et pieds)
- Nanisme micromélique : Atteinte de tous les segments de membres
Il existe de nombreux types de dysplasies
mais seulement une dizaine représentent 80 % des cas rencontrés.
- Importance de l’histoire familiale
- Importance de l’examen échographique complété par
le contenu utérin
Etude biométrique :
Tous les os doivent être mesurés. Le membre en question a une mesure < P5.
Différents rapports sont à
connaître :
- Fémur/Pied (N=0,99±0,6)
- Fémur/CA (N>0,20)
- Périmètre thoracique/Périmètre crânien (N=0,54
à 1,04) permet de suspecter une hypoplasie pulmonaire
Aspect du squelette :
Dysmorphie faciale :
Plusieurs paramètres permettent de typer la chondrodysplasie :
- Bosses frontales saillantes
- Ensellure nasale marquée
- Micrognathie
- Rétrognathie
- Ecart interorbitaire
Aspect des mains et
des pieds :
Trois aspects pouvant entrer dans les tableaux de chondrodysplasie :
-Agénésie
-Anomalies de segmentations
-Anomalies de position
Les dysplasies sont soit létales, soit compatibles avec la vie.
Dysplasies du squelette létales
Nanisme thanatophore
Isolé et décrit par
Maroteaux en 1967
Prévalence 0,3 à 0,7 pour 10 000
Mutation de type dominant
Risque de récurrence 2 %
Signes
principaux :
- Macrocéphalie (front proéminent)
- Thorax étroit ![]()
- Abdomen proéminent
- Membres courts, incurvés, boudinés



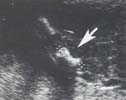
Signes associés :
- Aspect arqué des clavicules
- Hypoplasie des ailes iliaques
- Aspect en cloche de la cage thoracique
- Aplatissement des corps vertébraux
- Hydrocéphalie
- Hydramnios (70 % des cas)
- Hypominéralisation des vertèbres
En fait, deux sous groupe en fonction de la forme du crâne et du fémur :
Type 1 : Cas sporadique sans risque
de récidive
- Fémurs courbes en forme de combiné téléphonique
- Crâne normal
Type
2 : Correspondrait à une forme récessive autosomique
- Membres très courts mais droits
- Crâne en forme de trèfle

Achondrogénèse
Prévalence 1/40 000 à
1/80 000 naissances
Chondrodysplasie létale la plus sévère
La taille à la naissance varie de 25 à 35 cm
Signes
:
- Micromélie extrême ![]()
- Tronc court et large
- Thorax petit
- Macrocrânie constante
- Hydramnios et anasarque (50 %)
- Diminution constante de l’échogénicité du rachis
et du bassin (rachis échographiquement absent dès le 1er trimestre)
- Clarté nucale ou hygroma kystique fréquents (diagnostic possible
dès 12 SA)
Deux formes :
Type IA :
- Sévère et rare (20 %)
- Mode autosomique récessif
- Défaut complet d’ossification des os de la
base du crâne et des vertèbres
![]()
- Atteinte diffuse de l’ensemble des os longs ![]()
- Fractures du grill costal
Type IB :
- Sporadique 80 % des cas
- Pas de fracture
- Absence d’ossification vertébrale moins marquée
Ostéogénèse imparfaite
Rassemble un ensemble d’affections
différentes par leur aspect clinique et leur mode de transmission avec
en commun une anomalie du collagène, responsable d’une fragilité
des os et de fractures répétées.
Fréquence : 1/20 000 à 1/60 000 naissances
Signes échographiques
:
1. RCIU
2. Hydramnios
3. Anomalies du pôle céphalique :
- Hydrocéphalie
- Macrocéphalie
- Déformation céphalique spontanée ou provoquée
par la pression de la sonde
- Amincissement et faible échogénicité de la voûte
osseuse
- Déflexion céphalique maximale
4. Anomalies thoraciques :
- Thorax petit et étroit
- Hypoéchogénicité osseuse (diminution de l’ombre
acoustique des côtes)
- Fractures et cals osseux ![]()
5. Anomalies des membres :
- Nanisme (évident et précoce dans le type 2, peu marqué
dans le type 1)
- Fractures et cals osseux
![]()
![]()
![]()
- Déformation marquée des membres sans aucune symétrie
- Hypoéchogénicité osseuse (diminution de l’ombre
acoustique des os longs)
Quatre types selon Sillence (1979) :
Type I :
- Transmission dominante
- Assez bon pronostic
- Nanisme modéré
- Fractures survenant après la naissance, parfois au cours du 3ème
trimestre
- Sclérotiques bleues
Type II :
- Transmission récessive
- Constamment létale
- Nanisme sévère
- Fractures et cals osseux nombreux
- Sclérotiques bleues
- Déformations majeures des membres
Type III :
- Soit dominant, soit récessif
- Déformation progressive et sévère des membres et du rachis
post-natale
- Sclérotiques bleues (le bleu s’atténue avec l’âge)
Type IV :
- Autosomique dominant
- Forme la plus modérée
- Taille normale
- Slérotiques blanches
- Manifestation atténuée de la maladie
Les formes tardives à révélation post natale constituent la maladie de Lobstein. (maladie des « os de verre »), autosomique dominante (types I et IV)
Nanisme campomélique
1/200 000 naissances
Signes échographiques
:
- Nanisme mésomélique
- Incurvation concave et marquée des os longs
![]()
![]() (surtout des membres inférieurs) de façon symétrique
contrairement à l’ostéogenèse imparfaite.
(surtout des membres inférieurs) de façon symétrique
contrairement à l’ostéogenèse imparfaite.
- Thorax étroit
- Anomalies faciales à type de microrétrognathisme, d’hypertélorisme
et de division palatine parfois
- Pieds bots varus équins bilatéraux fréquents
- Anomalies viscérales (cardiaques et rénales) souvent associées
Hypophosphatasie
1/100 000
Transmission autosomique récessive
Anomalie du métabolisme phosphocalcique d’origine génique
provoquant un défaut de la minéralisation osseuse avec déficience
des phosphatases alcalines.
Signes échographiques :
- Très faible minéralisation du squelette
- Fractures in utero possibles
- Diffère de l’ostéogenèse imparfaite par la finesse
des extrémités osseuses
- Thorax étroit
- Micromélie variable
Dysplasies du squelette non létales
Achondroplasie
Chondrodysplasie non létale
la plus fréquente.
Prévalence : 1 cas sur 66000 naissances
Transmission autosomique dominante mais le plus souvent fait suite à
une néo-mutation liée à l’âge du père.
Signes échographiques:
- Nanisme micromélique à prédominance rhisomélique
(d’apparition tardive avec cassure de la courbe de croissance fémorale
après 26 SA)
- Fémurs courbes avec extrémités effilées
en biseau ![]()
- Anomalies possibles des mains avec brachydactylie ou écart anormal
entre les 3ème et 4ème doigts (aspect en trident)
- Macrocéphalie, face massive avec front bombant
![]() , ensellure nasale marquée en coup de hache
, ensellure nasale marquée en coup de hache ![]() , lèvres charnue, langue souvent protruse
, lèvres charnue, langue souvent protruse
La forme hétérozygote est compatible avec la vie.
Dysplasie thoracique asphyxiante de Jeûne
Transmission autosomique récessive
- Micromélie modérée
à prédominance proximale
- Thorax très étroit à côtes courtes
- Polydactylie absente
- Hypoplasie pulmonaire (70 %) fait le diagnostic
Chondrodysplasie ponctuée
Incidence : 0,9/100 000
Micromélie à
prédominance proximale et souvent asymétrique.
Présence de calcifications au voisinage des cartilages
de conjugaison. ![]()
Biopsie de trophoblaste : Déficit en enzymes peroxysomiales.
Type I :
- Mode dominant lié
à l’X
- Non rhizomélique
- Dysmorphie faciale fréquente
- Cataracte et pathologie cutanée présente dans 30 % des cas
- Cardiopathie et uropathie possible
Type II :
- Mode autosomique récessif
- Rhizomélique
- Anomalies marquées
- Les calcifications épargnent le rachis
- Retard psychomoteur sévère
Polydactylies
Se caractérisent
par la présence de doigts surnuméraires, le plus souvent petits
et malformés.
Les formes les plus fréquentes et en général
isolées sont post-axiales
![]()
![]() , siégeant sur le bord ulnaire de la main et fibulaire de la jambe.
, siégeant sur le bord ulnaire de la main et fibulaire de la jambe.
Les polydactylies pré-axiales
![]() sont plus rares, siégeant sur le bord interne du membre touché
et appartenant le plus souvent à des tableaux polymalformatifs.
sont plus rares, siégeant sur le bord interne du membre touché
et appartenant le plus souvent à des tableaux polymalformatifs.
Leur pronostic dépend de leur caractère isolé ou non. Certains syndromes plus fréquents comportant une polydactylie sont bien répertoriés.
Syndrome polydactylie-côtes courtes
Transmission autosomique récessive.
- Polydactylie
- Nanisme micromélique
- Thorax étroit avec côtes très courtes (coup de hache axillaire)
- Microcéphalie sévère
- Anomalies viscérales inconstantes (cardiaque, urinaire ou digestif)
- L’hypoplasie pulmonaire fait le diagnostic
Dysplasie chondro-ectodermique (syndrome d’Ellis et Van Creveld)
Rare, de transmission autosomique récessive, viable une fois sur deux.
- Polydactylie
- Etroitesse du thorax moins marquée
- Malformation cardiaque (50 %) des cas
Syndrome de Meckel-Gruber
Transmission autosomique récessive, létal.
Triade :
- Encéphalocèle
- Polydactylie
- Dysplasie rénale
Triade non obligatoirement complète.
Association possible :
- Microcéphalie
- Microphtalmie
- Cardiopathie
- Syndactylie
Syndactylies
Se caractérisent
par la soudure partielle ou complète de deux ou plusieurs doigts
![]()
![]() . Suspectée devant l’absence de motilité indépendante
des doigts. De nombreuses pathologies intègrent cette malformation.
. Suspectée devant l’absence de motilité indépendante
des doigts. De nombreuses pathologies intègrent cette malformation.
Syndrome d’Apert
Secondaire à une néo-mutation, s’observerait surtout en cas d’âge paternel avancé.
Associe :
- Syndactylie constante sévère et souvent
osseuse réalisant une « main en moufle »
![]()
![]()
![]()
- Turricéphalie (secondaire à une crâniostose
antérieure avec front très haut)
![]()
- Fente palatine fréquente
- Cardiopathie
- Hypoplasie pulmonaire
- Polykystose rénale
- Retard mental constant
Syndrome de Fraser
- Syndactylie peu sévère
- Malformation des organes génitaux externes
- Agénésie rénale bilatérale parfois
- Cryptophtalmie
- Retard mental
Nécessite un caractère figé, persistant de positions anormales avec mobilisation en bloc de deux segments de membre contigus adoptant une angulation anormale. En fin de grossesse, le diagnostic est beaucoup plus difficile.
Pied bot
Anomalie positionnelle la plus fréquente.
Embryologiquement, les pieds sont jusqu’à la 11 SA en adductus
équinovarus. La position définitive n’est observée
qu’après une rotation interne des membres inférieurs entre
10 et 12 SA. Les pieds bots malformatifs sont le plus souvent lés à
une anomalie de cette rotation. Leur diagnostic est difficile avant 14-15 SA.
De plus un pied bot peu apparaître plus tardivement.
La coupe passant par l’axe jambier se prolonge non pas par un profil du pied mais par un aspect de voûte plantaire légèrement déformée.
Le problème est de
différencier une simple anomalie positionnelle d’une malformation
vraie. Trois signes orientent vers un pied bot malformatif :
- Talon déshabité, l’aspect échogène du calcanéum
est remplacé par une cupule graisseuse
- La plante regarde en dedans et en arrière
![]()
- La malrotation reste fixée lors des mouvements
du membre inférieur
![]()
Les pieds bots sont le
plus souvent isolés mais doivent faire rechercher :
- Un problème neurologique (spina-bifida) ![]()
- Une polymalformation
- Certains nanismes
- Une anomalie osseuse de la jambe
- Une anomalie chromosomique en particulier trisomie 18
Les pieds bots bilatéraux doivent faire rechercher des anomalies positionnelles des mains (mains botes ou mains crispée) ou d’autres anomalies associées, en particulier cardiaque qui peuvent orienter vers une anomalie chromosomique.
Un pied bot unilatéral peut être associé à une agénésie tibiale.
On peut rapprocher des pieds
bots certaines anomalies mineures :
- Déviation en coup de vent des orteils par rapport aux métatarsiens
- Aspect de pied en piolet avec saillie des talons


Mains botes
Forment
une angulation anormale permanente irréductible par rapport aux avant-bras
qui peut atteindre ou dépasser 90°
![]()
![]() . Elles peuvent s’associer à des hémi-aplasies latérales
(surtout aplasie radiale).
. Elles peuvent s’associer à des hémi-aplasies latérales
(surtout aplasie radiale).
Rarement isolée, elles doivent faire rechercher une anomalie chromosomique (notamment trisomie 13ou 18).
Mains crispées
Doigts
en flexion permanente plus ou moins accentuée avec chevauchement ou tendance
au chevauchement de l’index sur le 3ème doigt et du 5ème
sur l’annulaire
![]() . Elles sont assez caractéristiques de la trisomie 18.
. Elles sont assez caractéristiques de la trisomie 18.
7 - Syndrome d’Immobilisme fœtal
Secondaire à un oligoamnios, il est facilement reconnaissable, les malformations induites sont positionnelles.
Primitif,
il fait coexister un hydramnios et des raideurs articulaires
![]()
![]() , c’est une arthrogrypose soit isolée, soit entrant dans le cadre
d’une polymalformation létale récidivant dans 25 % des cas..
, c’est une arthrogrypose soit isolée, soit entrant dans le cadre
d’une polymalformation létale récidivant dans 25 % des cas..
Le diagnostic repose sur l’existence de déformation des membres avec raideur articulaire permanente associée à une inactivité fœtale retrouvée lors de plusieurs examens échographiques successifs.
Devant une arthrogrypose isolée,
on évoque :
- Une origine maternelle : myasthénie transmise
- Une origine fœtale : -Myopathie de Steinert
- Myopathie à bâtonnets
- Myéloméningocèle lombaire
- Infection virale
- Dégénérescence des cellules motrices des cornes antérieures
de la moelle
Devant une arthrogrypose
entrant dans un tableau polymalformatif, on évoque les COFS syndromes
(Cerebro-Oculo-Facio-Skeletal), autosomiques récessifs et létaux.
* Type I :
- Arthrogrypose
- Hydramnios
- Hypoplasie pulmonaire
- Œdème sous-cutané
- Anomalies de la face
* Type II :
- S’ajoute la microcéphalie
- La microphtalmie
- Une anomalie de la courbure vertébrale
- Des malformations rénales parfois
8 - Tératogénicité et anomalies de membres
-Thalidomide et acide rétinoïque
:
¤ Amputation de membre unique ou multiple
¤ Phocomélie des quatre membres parfois
- Acide valproïque : Anomalies des extrémités
- Warfarine : Chondrodysplasie ponctuée
• Période optimale
de visualisation : 15 à 26 SA
• Biométrie du fémur
• Comparer les membres et leurs différents segments
• Importance des antécédents
Il nous semble indispensable de mesurer au moins le fémur, et de voir et « comparer » les membres et leurs différents segments.
